|
|
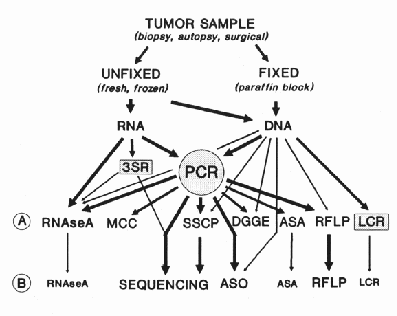
Due to their subtle nature, the point mutations are the most difficult to detect of all the genetic alterations. Nevertheless, there are quite a few techniques capable of recognising the presence of single base changes in mammalian genes. A diagram of the methods currently utilised for detection and characterisation of single base substitutions, deletions, and insertions is shown in figure 3.1. In the first row (A) are listed methods for detection of the presence of the mutation. In (B) are the methods that permit characterisation of the molecular nature of the mutation. While detection of the presence of a mutation might be sufficient for some diagnostic and prognostic purposes, characterisation of the single base substitutions is also important for several reasons.
The RNase A mismatch cleavage method is based on the ability of pancreatic ribonuclease to recognise and cleave single base mismatches in RNA:RNA [146] and RNA:DNA heteroduplexes [147]. The method is performed by hybridisation of the target sequence to a labelled complementary riboprobe, digestion with RNase A, and analysis of the resistant products by polyacrylamide electrophoresis in denaturing gels. Mutations are detected and localised by the presence and size of the RNA fragments generated by cleavage at the mismatches.
The single strand conformation polymorphism (SSCP) is another method developed by HAYASHI, SEKIYA, and colleagues [148, 149]. It is based on the differences in the secondary structure of single-strand DNA molecules differing in a single nucleotide, which also is frequently reflected in an alteration of their electrophoretic mobility in nondenaturing gel electrophoresis.
In denaturing gradient gel electrophoresis (DGGE), the double stranded DNA is subjected to electrophoresis in gels that have an increasing concentration of denaturant along the length of the gel [150]. The fragment melts while travelling through the gel. The melting proceeds in segments, called melting domains, because of the cooperative nature of the denaturation of the double-stranded DNA [151]. When a domain melts, the fragment assumes a branched structure that causes significant retardation of movement. Thus, the position of the fragment in the gel after a certain time of electrophoresis is determined by the history of melting of the fragment that is altered if the sequence is different. The principle of separation in DGGE is such that sequence changes in the melting domain of highest stability cannot be detected, because the fragment no longer has a branched structure when the last domain melts. If, however, a stretch of sequence that serves as an extremely stable domain is attached to one side of the fragment, then mutations at any sites within certain types of sequence context can be detected by DGGE [152]. This extra sequence of extremely high stability can be conveniently attached to the target sequence of PCR by using one primer that has 40 nucleotides of an artificial GC-rich sequence (GC-clamp) extending at its 5’-end. With the use of this clamp, DGGE may be able to detect nearly all possible mutations in any given sequences [153, 154].
Hybridisation with radioactively labelled allelic specific oligonucleotides (ASO) also has been applied to the detection of specific point mutations [155]. The method is based on the differences in the melting temperature of short DNA fragments differing by a single nucleotide. PCR has also generated exclusive methods for single point mutation detection, based on the selective amplification of specific alleles. The acronyms are numerous for a series of techniques based on the presence of mismatches at, or near, the 3’-end of the PCR primers. The mismatch between the primer and the wild-type sequence may impede the extension by the polymerase and little or no amplification occurs. On the other hand, the presence of a particular mutation, complementary to the primer, allows efficient amplification [156]. Variations of this approach are called allele specific amplification (ASA) [157, 158], amplification refractory mutation system (ARMS) [159], mismatched PCR [160], polymerase allele-specific amplification (PASA) [161], and mutation-specific PCR assay (MSPA) [162].
We successfully established an ARMS protocol (see section 3.3.1) for scanning two mutations.
© 2001 Alexander Binder