The standard method used to separate, identify, and purify DNA fragments is electrophoresis through agarose gels. The technique is simple, rapid to perform, and capable of resolving mixtures of DNA fragments that cannot be separated adequately by other sizing procedures. Furthermore, the location of DNA within the gel can be determined directly: Bands of DNA in the gel are stained with the intercalating dye ethidium bromide ; as little as 1 ng of DNA can be detected by direct examination of the gel in ultraviolet light[101].
Agarose, which is extracted from seaweed, is a linear polymer whose basic structure is shown in figure fig:agar.
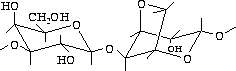
Figure 2.2: Agarose structure unit
Commercially available agarose is not completely pure, it is contaminated with other polysaccharides, salts, and proteins. These differences can affect both, the migration of the DNA and the ability of the DNA recovered from the gel to serve as a substrate to enzymatic reactions. Agarose gels are cast by melting the agarose in the presence of the desired buffer until a clear, transparent solution is achieved. The melted solution is then poured into a mold and allowed to harden. Upon hardening, the agarose forms a matrix, the density of which is determined by the concentration of the agarose. When an electric field is applied across the gel, DNA, which is negatively charged at neutral pH, migrates toward the anode. The electrophoretic migration rate of DNA through agarose gels is dependent upon four main parameters, which will be discussed below.
The molecular size of the DNA. Molecules of linear, duplex DNA, which are believed to migrate in an end-on position[102, 103] travel through gel matrices at rates that are inversely proportional to the logarithm of their molecular weights[104].
The agarose concentration. A DNA fragment of given size migrates at
different rates through gels containing different concentrations of agarose.
There is a linear relationship between the logarithm of the electrophoretic
mobility of DNA ( ![]() ) and gel concentration (
) and gel concentration ( ![]() ), which is described by
the equation:
), which is described by
the equation:
![]()
where ![]() is the free electrophoretic mobility and K
is the free electrophoretic mobility and K ![]() is the
retardation coefficient, a constant that is related to the properties of the
gel and the size of the migrating molecules. Thus, by using gels of different
concentrations, it is possible to resolve a wide-range of DNA fragments.
is the
retardation coefficient, a constant that is related to the properties of the
gel and the size of the migrating molecules. Thus, by using gels of different
concentrations, it is possible to resolve a wide-range of DNA fragments.
The conformation of the DNA. Closed circular, nicked circular and linear DNA of the same molecular weight migrate through agarose gels at different rates. The relative mobilities of the three forms are dependent primarily on the agarose concentration in the gel but are also influenced by the strength of the applied current, the ionic strength of the buffer, and the density of superhelical twists in the DNA.
The applied current. At low voltages, the rate of migration of linear DNA fragments is proportional to the voltage applied. However, as the electric field strength is raised, the mobility of high-molecular-weight fragments of DNA is increased differentially. Thus, the effective range of separation of agarose gels decreases as the voltage is increased. Gels should be run at no more than 5 V/cm.
Base composition and temperature. The electrophoretic behavior of DNA in agarose gels (by contrast to polyacrylamide gels[105]) is not significantly affected either by the base composition of the DNA[106] or the temperature at which the gel is run. Thus, in agarose gels the relative electrophoretic mobilities of DNA fragments of different sizes do not change between 4psy176 C and 30psy176 C.

Figure 2.3: Photograph of agarose gel, showing taped edges and well-forming comb
in position.
The agarose concentration is varied for different fragment ranges. For
analyzing the complete 2ar codon (1239 bp), a 1% agarose gel ![]() is made by dissolving agarose in 1
is made by dissolving agarose in 1 ![]() TAE buffer by heating in a
microwave oven. After cooling to about 60psy176 C, ethidium
bromide
TAE buffer by heating in a
microwave oven. After cooling to about 60psy176 C, ethidium
bromide ![]() is added
to a final concentration of 0.5
is added
to a final concentration of 0.5 ![]() g/ml. The agarose solution is poured into
a taped gel former mold to make the gel. A well-forming comb (12 slots for
minigels) is placed near one edge of the gel. The gel is cooled to harden until
it becomes milky and opaque (approximately one hour). The gel mold is placed
horizontally into the electrophoresis tank, which is filled with 1
g/ml. The agarose solution is poured into
a taped gel former mold to make the gel. A well-forming comb (12 slots for
minigels) is placed near one edge of the gel. The gel is cooled to harden until
it becomes milky and opaque (approximately one hour). The gel mold is placed
horizontally into the electrophoresis tank, which is filled with 1 ![]() TAE
TAE
![]() (0.5
(0.5 ![]() g/ml ethidium
bromide).
g/ml ethidium
bromide).
The gel loading buffer is applied to the samples and they are carefully added
to individual wells![]() . The
electrophoresis is run by 70-100 V/20-80 mA for about an hour or at 20 to 30
V overnight. The size of fragments can be determined by calibrating the gel,
using known standards (e.g.,
. The
electrophoresis is run by 70-100 V/20-80 mA for about an hour or at 20 to 30
V overnight. The size of fragments can be determined by calibrating the gel,
using known standards (e.g., ![]() DNA EcoRI / HindIII
digest, Boehringer Mannheim, or 100bp ladder, BioVentures, Inc.), and comparing the
distance the unknown fragment has migrated.
DNA EcoRI / HindIII
digest, Boehringer Mannheim, or 100bp ladder, BioVentures, Inc.), and comparing the
distance the unknown fragment has migrated.
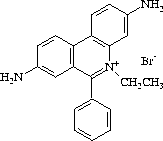
The most convenient method of visualizing DNA in agarose gels is by use of the fluorescent dye ethidium bromide [101] (2,7-Diamino-10-ethyl-9-phenyl-phenanthridinium bromide). This substance (see figure fig:etbr) contains a planar group that intercalates between stacked bases of DNA. The fixed position of this group and its close proximity to the bases causes dye bound to DNA to display an increased fluorescent yield compared to dye in free solution. UV-irradiation absorbed by the DNA at 260 nm and transmitted to the dye, or irradiation absorbed at 300 nm and 360 nm by the bound dye itself, is emitted at 590 nm in the red-orange region of the visible spectrum.
Ethidium bromide can be used to detect both single- and double-stranded nucleic acids. However, the affinity of the dye for single-stranded nucleic acid is relatively low and the fluorescent yield is poor.